During the Christmas holidays I released a new version of ProbABEL (v0.4.2). The official release announcement can be found here. ProbABEL is a toolset that allows running GWAS (Genome-Wide Association Studies) in a fast and efficient manner. It implements regression using the linear, logistic or Cox proportional hazards models.
This version is mostly a bug fix release. The most important user-visible change is the fact that the ‘official’ name for the wrapper script that runs a GWAS over a range of chromosomes is now called probabel instead of probabel.pl. This change was induced by my attempts to get ProbABEL packaged in the Debian Linux repositories. One of the warnings that occurred during the package creation process was a Lintian warning that said that scripts with ‘language extensions’ are not allowed. There are several reasons for that, but the one I found most compelling was the fact that the user shouldn’t be concerned with the programming/scripting language we used to write it in. Moreover, being ‘agnostic’ in this matter also allows us to write such a script in a different language.
Of course, we have left the original name in place (via a symlink) in order not to disrupt any current pipelines. If the user runs the script with the old name a warning appears, urging him/her to start using the new name and that the old name will be deprecated in the future.
In the mean time, ProbABEL v0.4.1 has been accepted in Debian (unstable) and as of today it is also available in Debian ‘testing’. Lots of thanks to the Debian Med team that helped me a lot in preparing the .deb package. Note that the package has been split up in probabel (architecture-dependent files) and probabel-examples (with architecture independent files: the examples). See the Debian Package Tracking System page for ProbABEL for more details of the package.
From Debian the package has trickled down to Ubuntu as well (Launchpad page here), so it will be available by default in the next Ubuntu release (14.04, a.k.a. Trusty Tahr).
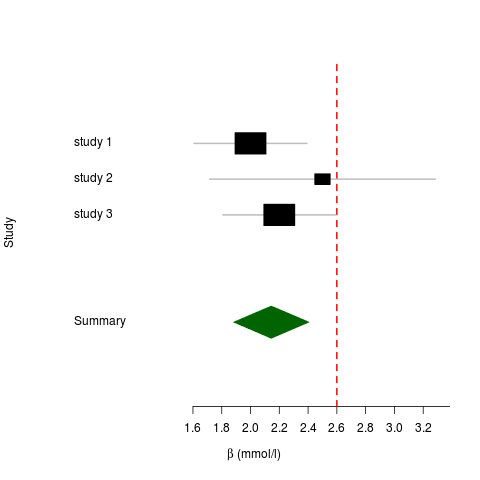
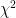 values in the output files. At the moment these are based on the LRT (likelihood ratio test), except where that doesn’t make sense (e.g. when using the
values in the output files. At the moment these are based on the LRT (likelihood ratio test), except where that doesn’t make sense (e.g. when using the